![]()
from the 2021 ISB Virtual Microbiome Series
Let's get the slides first (use your computer, phone, TV, fridge, anything with a 16:9 screen)
https://gibbons-lab.github.io/isb_course_2021/16S

💻 Let's switch to the notebook and get started
Click me to open the notebook!

All output we generate can be found in the treasure_chest folder at
https://github.com/gibbons-lab/isb_course_2021
or materials/treasure_chest in the Colaboratory notebook.
Pronounced like wind chime.
Created ~2010 during the Human Microbiome Project (2007 - 2016) under the leadership of Greg Caporaso and Rob Knight.
QIIME 2 is a powerful, extensible, and decentralized microbiome analysis package with a focus on data processing and analysis transparency.
Quantitative Insights into Microbial Ecology
Essentially, QIIME is a set of commands to transform microbiome data into intermediate outputs and visualizations.

It's commonly used via the command line.
QIIME 2 was introduced in 2016 and improves upon QIIME 1, based on user experiences during the HMP.
Major changes:
- integrated tracking of data provenance
- semantic type system
- extendable plugin system
- multiple user interfaces (in progress)
QIIME 2 comes with a lot of help, including a wide range of tutorials, general documentation and a user forum where you can ask questions.
QIIME 2 manages artifacts, which are basically intermediate data that feed into actions to either produce other artifacts or visualizations.


Artifacts often represent intermediate steps, but Visualizations are end points meant for human consumption ☝️.


The 16S gene is universal and contains interspersed conserved regions perfect for PCR priming and hypervariable regions with phylogenetic heterogeneity.
Photo by Nathan Jennings.


- 15 samples from 5 environments
- honey bee gut, cenotes in Yucatan (freshwater), ocean, human gut, soil
Photos by Dmitry Grigoriev, Jared Rice, Matt Hardy, Alex Block, and Roman Synkevich.


@SRR2143527.13917 13917 length=251
TACGTAGGTGGCGAGCGTTATCCGGAATTATTGGGCGTAAA...
+
BBBBAF?A@D2BEEEGGGFGGGHGGGCFGFHHCFHCEFGGH...
We have our raw sequencing data, but QIIME 2 only operates on artifacts. How do we convert our data into an artifact??
🐥 or 🥚?
💻 Let's switch to the notebook and get started
We will now run the DADA2 plugin, which will do 3 things:
- filter and trim the reads
- find the most likely original sequences in the sample (ASVs)
- remove chimeras
- count the abundances
- trim low quality regions
- remove reads with low average quality
- remove reads with ambiguous bases (Ns)
- remove PhiX (bacteriophage genome commonly added as a control to sequencing runs)

Expectation-Maximization (EM) algorithm used to build a dataset-specific error model and find true amplicon sequence variants (ASVs), all at once.

The primers used in this study were F515/R806. The numbers denote positions along the 16S gene. So, how long is the amplified fragment?
We now have a table containing the counts for each ASV in each sample. We also have a list of ASVs.
:thinking_face: Do you have an idea for what we could do with these two data sets? What quantities might we be interested in?
In microbial community analysis we are usually interested in two different families of diversity metrics, alpha diversity (ecological diversity within a sample) and beta diversity (ecological differences between samples).
How diverse is a single sample?

- richness: how many taxa do we observe (richness)?
→ total number of observed taxa - evenness: how evenly are abundances distributed across taxa?
→ Evenness index - mixtures: metrics that combine both richness and evenness
→ Shannon index, Simpson's Index
Alpha diversity will provide a single value/covariate for each sample.
It can be treated as any other sample measurement and is suitable for classic univariate tests (t-test, Mann-Whitney U test).
How different are two or more samples/donors/sites from one another other?

- unweighted: how many taxa are shared between samples?
→ Jaccard index, unweighted UniFrac - weighted: do shared taxa have similar abundances?
→ Bray-Curtis distance, weighted UniFrac
Do samples share genetically similar taxa?

Weighted UniFrac further scales phylogenetic branch lengths by abundances.
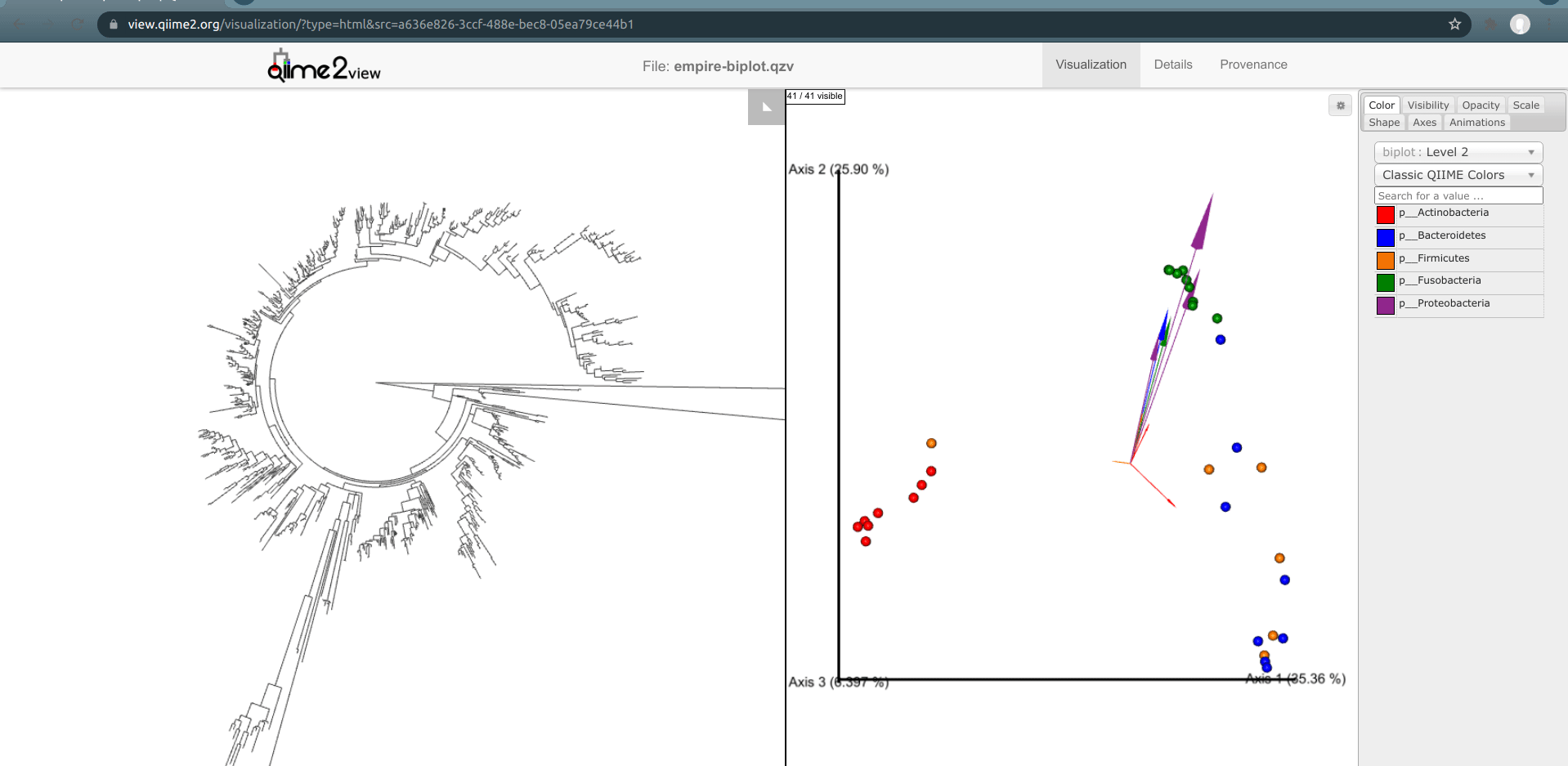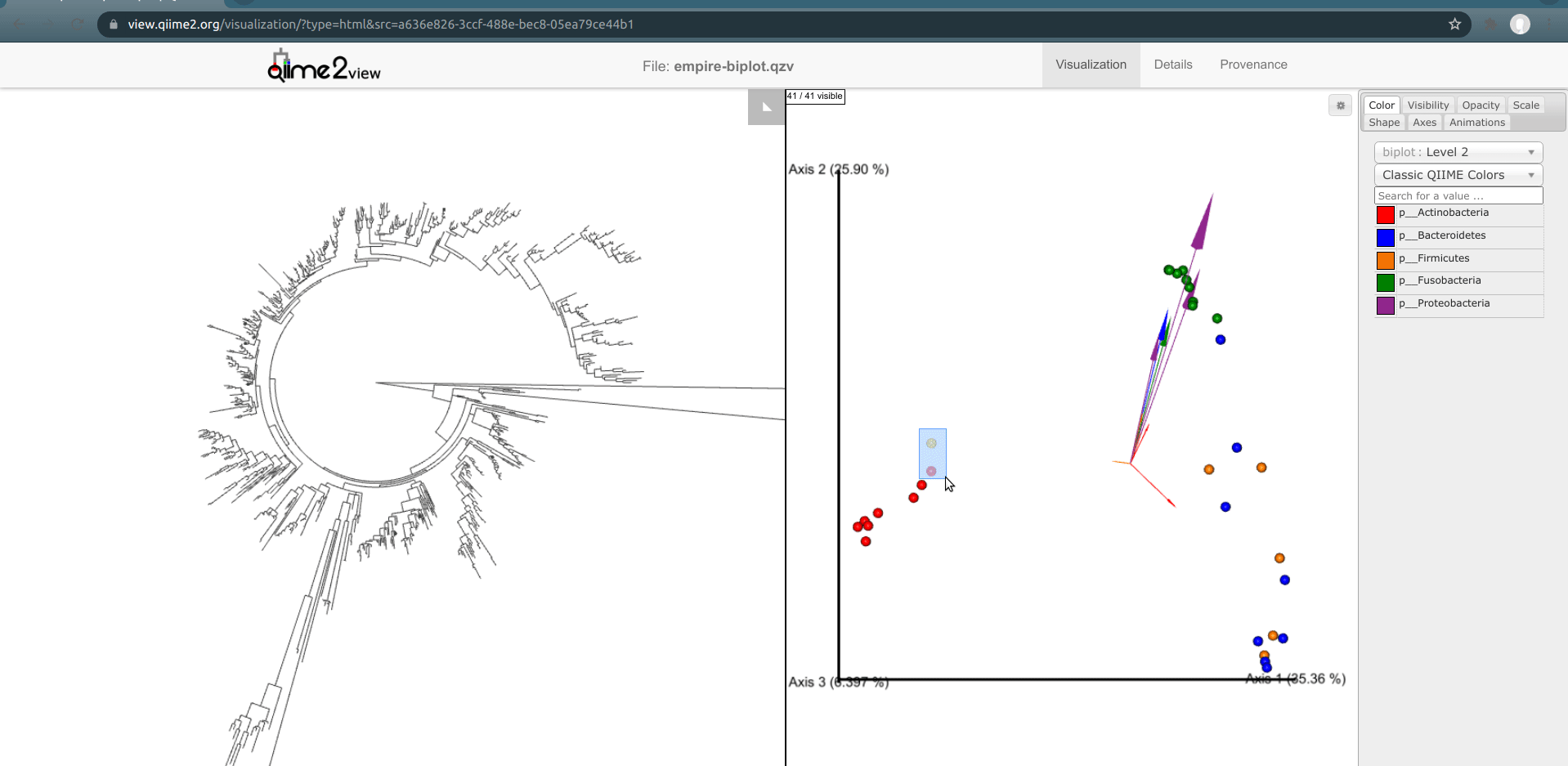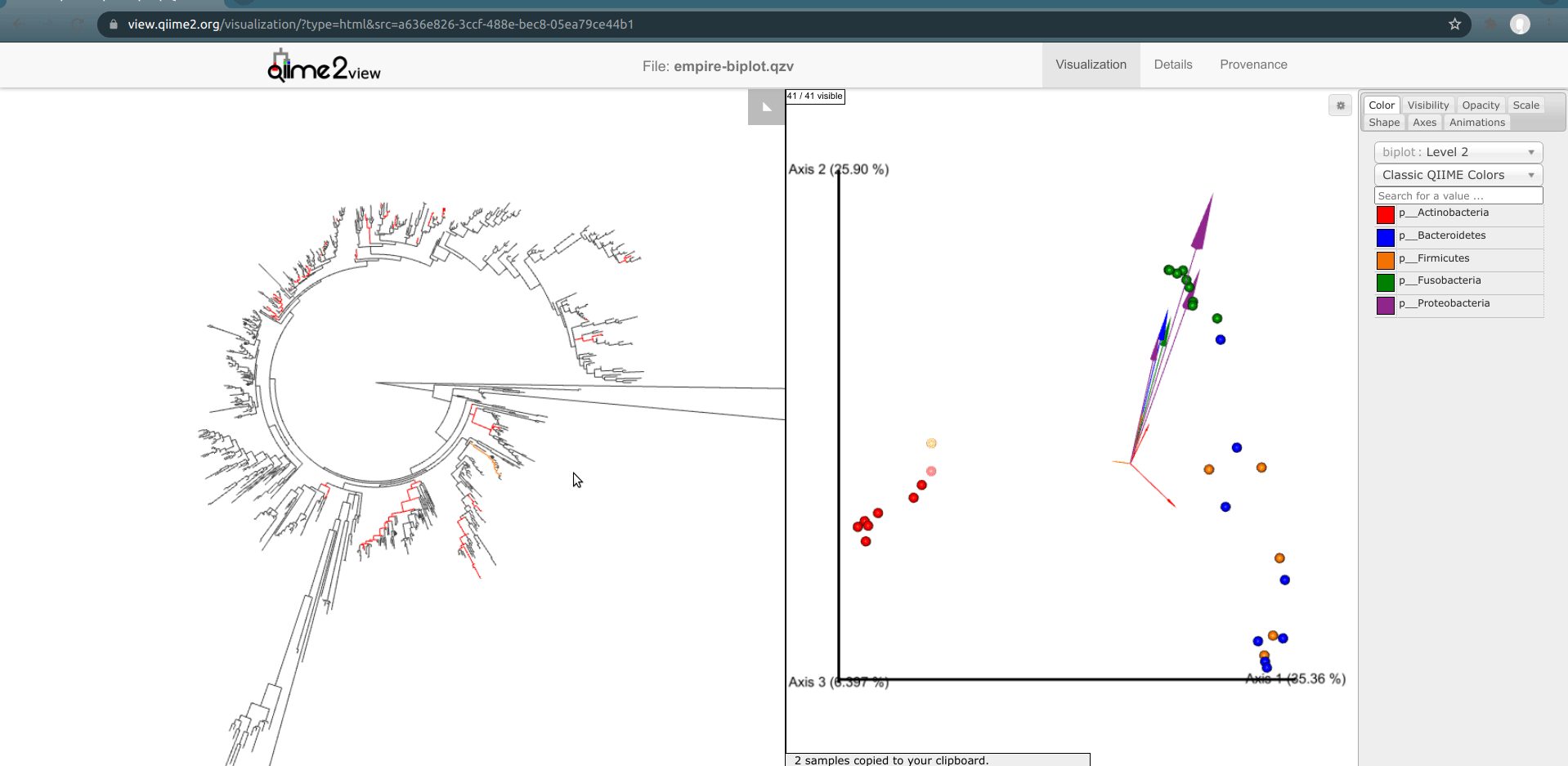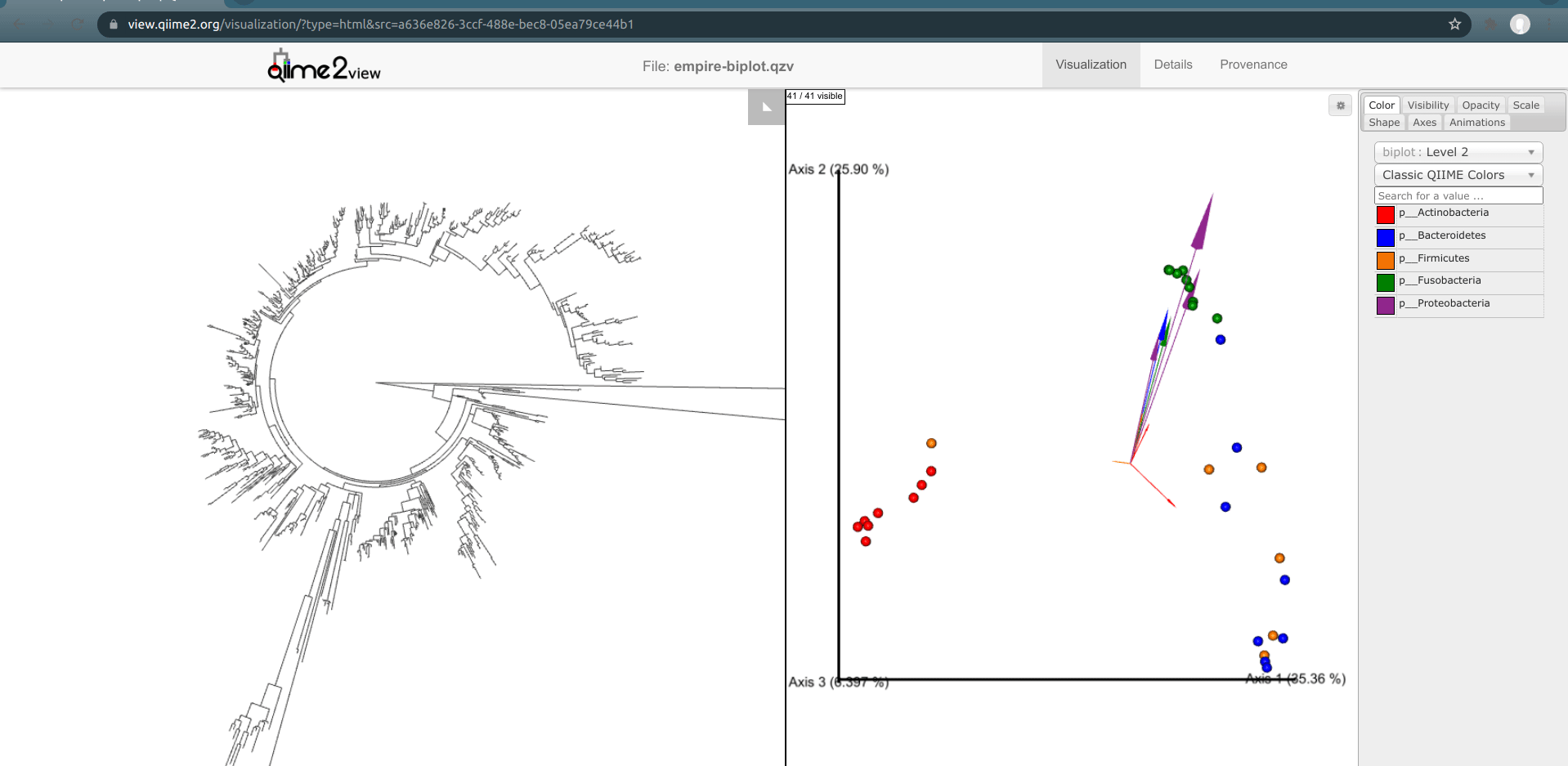
One of the basic things we might want to look at is how the ASVs across all samples are evolutionarily related to one another. That is, we are often interested in their phylogeny.
Phylogenetic trees are built from multiple sequence alignments and sequences are arranged by sequence similarity (branch length).
We can visualize this tree with EMPRESS.

More complicated. Usually not normal and very heterogeneous. PERMANOVA can deal with that.

💻 Let's switch to the notebook and calculate the diversity metrics
We are still just working with sequences and we have no idea what organisms those sequences correspond to.
:thinking_face: What would you do to go from a sequence to an organism's name?

Even though directly aligning our sequences to a database of known genes seems most intuitive, this does not always work well in practice. Why?

Instead, use subsequences (k-mers) and their counts to predict the lineage/taxonomy with machine learning methods. For 16S amplicon fragments, this approach often provides better generalization and faster results.
💻 Let's switch to the notebook and assign taxonomy to our ASVs
Are certain taxa only found in one environment? Are others more widely distributed?


Note:
Welcome to the 2021 ISB Microbiome Project challenge. Create a figure submission in this channel for a chance to win an awesome ISB T-shirt. Our team will pick one winning submission for each geographical region.
Rules:
- one entry per participant
- figure content has to be created only using Qiime 2 and the EMP data set from the course
- 4 panels (sub-figures) maximum, a single figure/plot is perfectly okay
- must include text that provides the region you identify with and a caption for the figure
Regions: Regions are from the United Nations Geoscheme. You can use the map in the link with the following changes:
- North America is split into: North America, United States, and Canada
- Antarctica is included as a region