Jobs
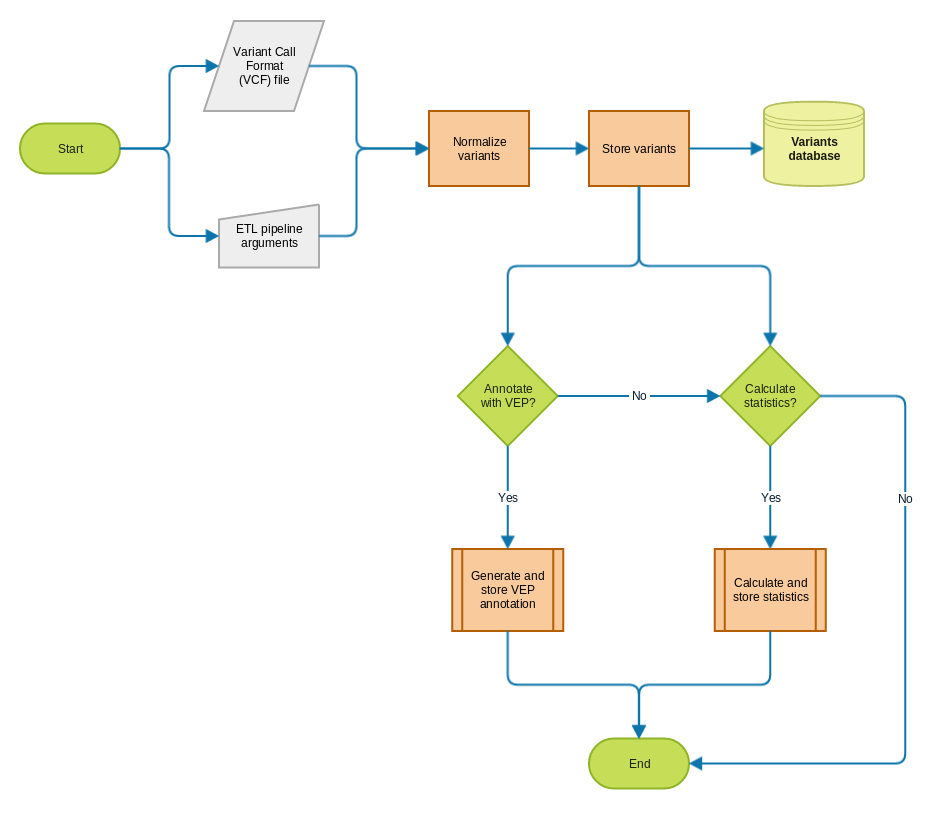
The main goal of the EVA pipeline is to store variants read from VCF files, calculate population statistics and annotate them using Ensembl's Variant Effect Predictor.
In order to do so, the following jobs are available:
- Initializing the database (work in progress)
- Processing a genotyped VCF
- Processing an aggregated VCF
- Re-annotating variants (work in progress)
- Re-calculating statistics (work in progress)
Please note this section is a work in progress and more details about the structure of each job will be added in the future.
It is not yet necessary to run this job in order to have a ready-to-use database. In the future, some preparation will be required for improved efficiency when querying the database.
This preparation will at least involve loading a feature set for the genome assembly the variation data is based on, to allow to translate from gene/transcript name to genomic coordinates.
One of the sources of data for the EVA ETL pipeline is a Variant Call Format (VCF) file listing the sample genotypes for each variants.
First, the alleles from columns REF and ALT are normalized to ensure that different representations across multiple VCFs will be recognized as the same. More information about the normalization process can be found in this presentation. Once normalized, variants are stored into the database (currently MongoDB).
Population statistics and annotation from Ensembl VEP can optionally be generated and stored along with the core variant information. These processes are shared across multiple jobs (VCF processing, re-annotation, etc) and will be documented separately.
TODO
TODO
TODO